We have recently demonstrated that the ancient kinases ULK1 and ULK2, which were thought to primarily stimulate autophagy initiation, have distinct functions in skeletal muscle. Our results revealed that ULK2 is enriched in skeletal muscle and that its deficiency impaired the degradation of insoluble ubiquitinated protein aggregates, thereby compromising proteostasis and contractile function. Furthermore, our studies demonstrated that ULK2 stimulates the recognition and proteolytic sequestration of these protein aggregates in association with the chaperones p62 and NBR1. Of note, these findings were not replicated by ULK1 deficiency. Current studies in the lab are dissecting the mechanisms involved in ULK2 function and exploring its therapeutic potential in several myopathies and aging-related muscle dysfunction.
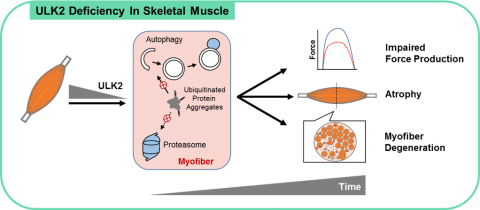
Diagram shown at Fuqua et al, FASEB J 2019.
Autophagy plays an instrumental role in exercise-induced adaptations in skeletal muscle. Impairments in autophagy are common in chronic metabolic diseases (e.g., obesity and type II diabetes) and have detrimental effects on mitochondrial and contractile functions. We are particularly interested in understanding the molecular cross-talk between autophagy, or autophagy proteins, and other cellular pathways that are fundamental for metabolic homeostasis such as mitochondrial biogenesis, substrate selection/oxidation, as well as protein and organelle turnover. These studies have direct implications not only for obese and diabetic patients, but also for the preservation of muscle mass and function in conditions associated with muscle atrophy, such as immobilization and aging.
